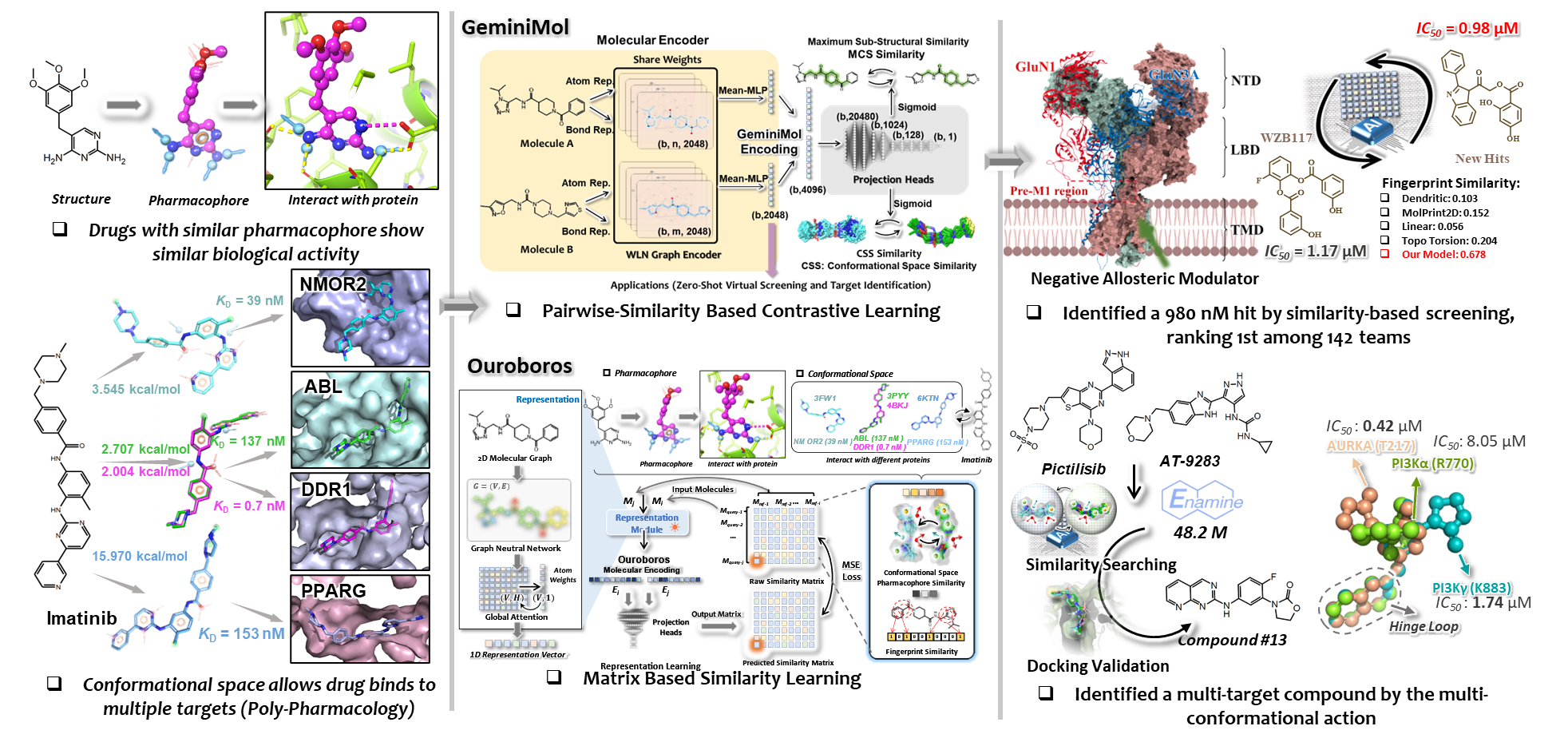
GeminiMol
Pairwise-similarity contrastive learning
GeminiMol learns from paired molecules, bringing encodings closer when their conformational and pharmacophore profiles are similar.
Validated in similarity search and screening.
Molecular representation · Drug discovery
We develop computational methods for molecular representation, property prediction, and structure generation. Ouroboros is one recent effort within a broader program that also studies protein–ligand interactions and cellular phenotype modeling.
Postdoctoral Researcher
Institute of Systems Medicine
Chinese Academy of Medical Sciences
A GNN maps molecular graphs to 1D vectors shaped by molecular fingerprint, conformational-space, and pharmacophore similarity.
Independent property decoders provide objectives for updating molecular encodings.
An independent Transformer decodes modified vectors to SMILES for subsequent prioritization and evaluation.
Selected research
Advanced Science · 2026
Ouroboros examines how a molecular representation foundation can be extended into a generation engine. A similarity-matrix objective organizes an encoding space informed by molecular fingerprints, conformational space, and pharmacophores; task-specific property decoders and an autoregressive SMILES decoder then support prediction, search, and iterative optimization.
GeminiMol and Ouroboros both use conformational-space and pharmacophore similarity as chemical priors. The later model changes how those relationships supervise the representation space.
GeminiMol
GeminiMol learns from paired molecules, bringing encodings closer when their conformational and pharmacophore profiles are similar.
Validated in similarity search and screening.
Ouroboros
Ouroboros supervises batches with full molecular similarity matrices, allowing multiple relative relationships to organize the encoding space together.
Validated in prediction, guided generation, and molecular discovery.
01 · Encode
A global-attention GNN maps each molecular graph to a 2048-dimensional vector. Training against molecular similarity matrices integrates fingerprint, conformational-space, and pharmacophore relationships.
02 · Navigate
Relevant property decoders translate encodings into task-specific predictions and provide objectives for navigating the representation space.
03 · Decode
An autoregressive Transformer reconstructs SMILES from input or revised encodings, yielding candidate structures for subsequent prioritization and evaluation.
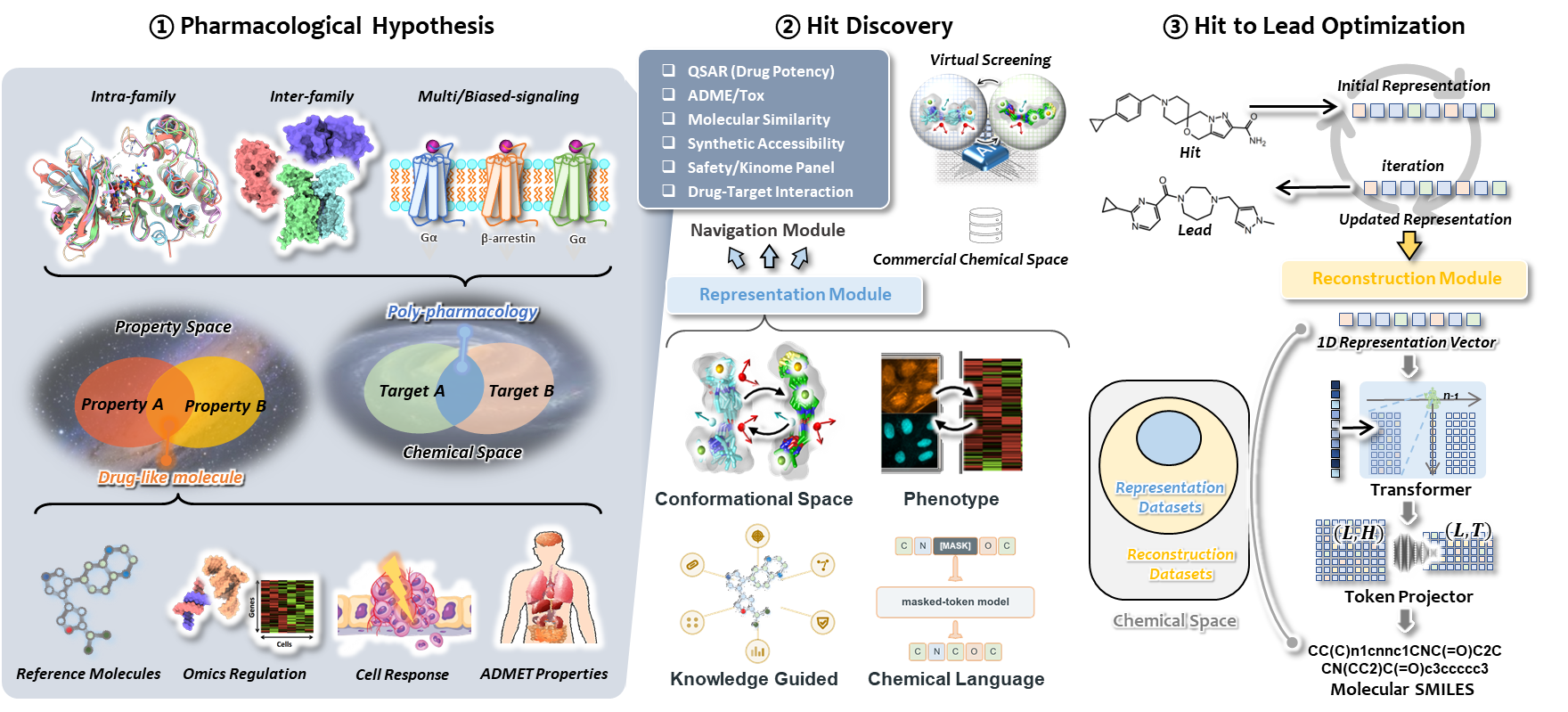
A pharmacological hypothesis defines what the system should optimize. A relevant property decoder turns the representation into a prediction for that objective; its signal guides movement through encoding space, and the SMILES decoder reconstructs revised encodings as candidate structures.
A desired profile, reference molecule, phenotype, or target combination.
A property decoder or similarity target expresses the task in the representation space.
Search or iterative updates move molecular vectors toward the objective.
The generative decoder returns candidate SMILES for subsequent evaluation.
After a hypothesis defines an objective, the same foundation can support complementary search strategies.
For virtual screening or de novo exploration, the objective ranks or guides candidate encodings across a broader chemical space.
Starting from a known hit, its encoding is iteratively updated toward the objective while proximity to the starting molecule can be monitored.
Several reference sets or objectives can be combined to examine candidates consistent with a multi-target hypothesis.
Related research
These projects address molecular representation, phenotype-based screening, protein structure and interactions, and synthetic accessibility. Together they provide context for the development of Ouroboros.
2026
Representation & generation
Property-guided molecular generation in a learned encoding space.
A shared representation for molecular graphs, property prediction, and SMILES generation.
2024
Molecular representation
Conformational-space profiling for general molecular representation.
Hybrid contrastive learning informed by conformational and pharmacophore similarity across distinct molecular scaffolds.
2025
Multimodal phenotype
Aligning molecular graphs with cellular morphology.
A foundation model aligning molecular graphs with cell images for phenotypic virtual screening.
2026
Protein conformations
Evaluating simulated protein conformational landscapes.
A benchmark for the diversity and plausibility of simulated protein conformational landscapes.
2022
Protein interactions
Combining structural and sequence motifs for protein-interaction analysis.
A co-driven computational method for PPI discovery, interaction modeling, and molecular-glue research.
2023
Synthetic accessibility
Predicting synthetic accessibility for molecular design.
A deep-learning model for identifying compounds likely to be difficult to synthesize.
Community work
Alongside published studies, we maintain practical tools that support database searching, molecular simulation, and reproducible computer-aided drug-design workflows.
01 · Database navigation
A browser-based navigator that sends one query to multiple biomedical databases, designed to make routine literature and data lookup more direct.
02 · Molecular dynamics
Automation for Desmond system setup and molecular-dynamics simulation, paired with a configurable pipeline for trajectory processing and analysis.
03 · CADD workflows
Shell utilities for virtual screening, cross-docking, and protein modeling with Schrödinger and Rosetta, shared to support reproducible computational workflows.
Recent publications
The full record contains 33 publications and book chapters from 2020–2026.
Loading publication record…
Research log
An archive of research releases, competition results, community tools, and publication milestones from the earlier site.
The Ouroboros study, Learned Conformational Space and Pharmacophore Into Molecular Foundational Model, was published in Advanced Science.
The PhenoModel study on multimodal phenotypic drug design was published in Acta Pharmaceutica Sinica B.
We introduced PhenoScreen, a dual-space contrastive learning framework combining conformational-space and molecular-image similarities for virtual screening.
Using GeminiMol, we identified an inhibitor of GluN1/GluN3A (IC50 = 0.98 μM); the work received first prize in the 2023 Shanghai International Computational Biology Innovation Competition.
The GeminiMol paper was published in Advanced Science.
We introduced GeminiMol, which incorporates conformational-space information into molecular representation learning; see the code and bioRxiv preprint.
DeepSA was published in the Journal of Cheminformatics; the DeepSA web server is also available.
Together with Shihang Wang, we developed DeepSA, a sensitive model for assessing the synthetic accessibility of small molecules.
We expanded the original plmd script and renamed it AutoMD, adding support for more force fields and customizable trajectory-analysis pipelines.
We developed the motif-driven PPI-Miner method. The study appeared in the Journal of Chemical Information and Modeling, with source code on GitHub.
We released a database of potential CRBN substrates, covering human proteins with a β-hairpin loop and a surface complementary to CRBN.
We identified an allosteric pocket on the SARS-CoV-2 spike protein and designed inhibitors intended to restrict its conformational change; the study was published in the Journal of Medicinal Chemistry.
We reported five candidate small-molecule antiviral blockers targeting the SARS-CoV-2 spike protein in Acta Pharmacologica Sinica.
We released automated Schrödinger workflows for cross-docking (XDock), virtual screening (GVSrun), and molecular-dynamics simulation (plmd), with the source code on GitHub.
We released Biodb-Search, a user-friendly biomedical database navigator intended to make literature research more efficient.
We released GetPDB, an automated script for downloading single-chain protein structures by UniProt ID.
Trajectory
Research training has progressed from life science and structural modeling to molecular, protein, and multimodal machine learning.
2019
Northeast Agricultural University
Biological foundations and early computational drug-discovery workflows.
2020–2024
ShanghaiTech University
Structural biology, molecular dynamics, protein interactions, and physically informed molecular representation.
Current
Institute of Systems Medicine, CAMS
Molecular and protein foundation models, phenotype-based learning, and translational drug-discovery applications.
Contact
We welcome discussions with researchers working across molecular modeling, machine learning, and experimental drug discovery.
Wanglin1102@outlook.com