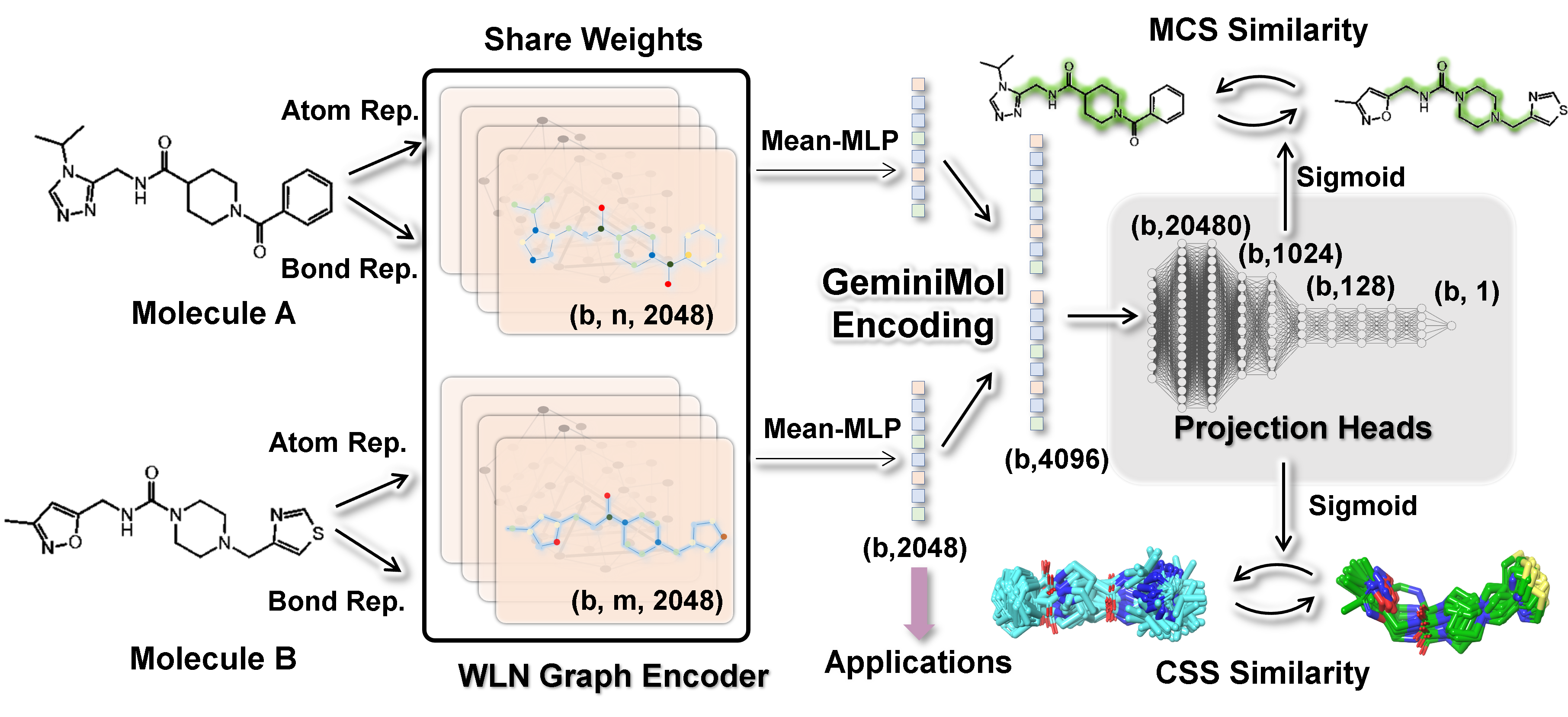
This repository provides the official implementation of the GeminiMol model, training data, and utilities. In this work, we propose a hybrid contrastive learning framework, which conducts inter-molecular contrastive learning by multiple projection heads of conformational space similarities (CSS). Please also refer to our paper for a detailed description of GeminiMol.
The molecular representation model is an emerging artificial intelligence technology for extracting features of small molecules. Inspired by the dynamics of small molecules in solution, introducing the conformational space profile into molecular representation models is a promising aim. The conformational space profile covers the heterogeneity of molecule properties, such as the multi-target mechanism of drug action, recognition of different biomolecules, dynamics in cytoplasm and membrane, which may facilitate further downstream application and generalization capability of molecular representation model.
PharmProfiler.py, which facilitates virtual screening and target identification.PropPredictor.py, which facilitates the deployment and repurposing of QSAR and ADMET prediction models.GeminiMol is a pytorch-based AI model. To set up the GeminiMol model, we recommend using conda for Python environment configuration. If you encounter any problems with the installation, please feel free to post an issue or discussion it.
Installing MiniConda (skip if conda was installed)
wget https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
sh Miniconda3-latest-Linux-x86_64.sh
Creating GeminiMol environment
conda create -n GeminiMol python=3.8.16
conda activate GeminiMol
Setting up GeminiMol PATH and configuration
git clone https://github.com/Wang-Lin-boop/GeminiMol
cd GeminiMol/
echo "# GeminiMol" >> ~/.bashrc
echo "export PATH=\"${PWD}:\${PATH}\"" >> ~/.bashrc # optional, not required in the current version
echo "export GeminiMol=\"${PWD}\"" >> ~/.bashrc
source ~/.bashrc
echo "export geminimol_app=\"${GeminiMol}/geminimol\"" >> ~/.bashrc # geminimol applications
echo "export geminimol_lib=\"${GeminiMol}/models\"" >> ~/.bashrc # geminimol models
echo "export geminimol_data=\"${GeminiMol}/data\"" >> ~/.bashrc # compound library
source ~/.bashrc
In this repository, we provide the pre-trained GeminiMol and CrossEncoder models.
Download model parameters and weights via Google Driver and HuggingFace
Here is an example of how to download a model from huggingface. Besides wget, you can also download the model directly from Google Cloud Drive or huggingface using your browser.
git clone https://huggingface.co/AlphaMWang/GeminiMol
Then, we need place the models to the ${GeminiMol}/models.
Download all chemical datasets via Zenodo for applications
cd ${geminimol_data}
wget https://zenodo.org/records/10450788/files/ChemDiv.zip # compound library for virtual screening
wget https://zenodo.org/records/10450788/files/DTIDB.zip # DTI database for target identification
for i in Benchmark*.zip css*.zip Chem*.zip;do
mkdir ${i%%.zip}
unzip -d ${i%%.zip}/ $i
done
unzip -d compound_library/ ChemDiv.zip
unzip -d compound_library/ DTIDB.zip
The expected structure of GeminiMol path is:
GeminiMol
├── geminimol # all code for GeminiMol and CrossEncoder
│ ├── model # code for GeminiMol and CrossEncoder models
│ │ ├── CrossEncoder.py # model and methods for CrossEncoder
│ │ ├── GeminiMol.py # model and methods for GeminiMol models
│ ├── utils # utils in this work
│ │ ├── chem.py # basic tools for cheminformatics
│ │ ├── dataset_split.py # tools for dataset split
│ │ ├── fingerprint.py # basic tools for molecular fingerprints
│ ├── Analyzer.py # analysis given molecules using presentation methods
│ ├── AutoQSAR.py # molecular proptery modeling by AutoGluon
│ ├── PropDecoder.py # molecular proptery modeling by our PropDcoder
│ ├── FineTuning.py # molecular proptery modeling by fine-tune GeminiMol models
│ ├── Screener.py # screening molecules by GeminiMol similarity
│ ├── CrossEncoder_Training.py # scripts for training the CrossEncoders.
│ ├── GeminiMol_Training.py # scripts for training the GeminiMol models.
│ ├── benchmark.py # benchmarking presentation methods on provide datasets
├── data # training and benchmark datasets in this work
│ ├── Benchmark_DUD-E # virtual screeening benchmark, optional
│ ├── Benchmark_LIT-PCBA # virtual screeening benchmark, optional
│ ├── Benchmark_TIBD # target identification benchmark, optional
│ ├── Benchmark_QSAR # QSAR and ADMET benchmarks, optional
│ ├── Chem_SmELECTRA # text backbone of chemical language, optional
│ ├── css_library # CSS training data, optional
│ ├── benchmark.json # dataset index for benchmark tasks, optional
│ ├── database.csv # molecular datasets in this work, optional
│ ├── compound_library # the compound librarys
│ │ ├── DTIDB.csv # dataset used in target identification
│ │ ├── ChemDiv.csv # library of common commercial compounds
│ │ ├── Specs.csv # library of common commercial compounds
├── models # CrossEncoder and GeminiMol models
│ ├── CrossEncoder # CrossEncoder, optional
│ ├── GeminiMol # GeminiMol, recommended for zero-shot tasks
Before running GeminiMol, you need to install the basic dependency packages.
Installing the RDkit for generating fingerprints
pip install rdkit
Installing the statatics and plot packages
pip install six
pip install oddt scikit-learn matplotlib scipy==1.10.1
Installing the dependency packages of GeminiMol
pip install torch==1.13.1+cu116 torchvision==0.14.1+cu116 \
--extra-index-url https://download.pytorch.org/whl/cu116
pip install dgl==1.1.1+cu116 -f https://data.dgl.ai/wheels/cu116/repo.html
pip install dglgo==0.0.2 -f https://data.dgl.ai/wheels-test/repo.html
pip install dgllife==0.3.2
If you intend to reproduce the benchmark results in our work, it is required to install the AutoGluon.
pip install autogluon==0.8.1 # requried for AutoQSAR
As a molecular representation model, GeminiMol finds applications in ligand-based virtual screening, target identification, and quantitative structure-activity relationship (QSAR) modeling of small molecular drugs.
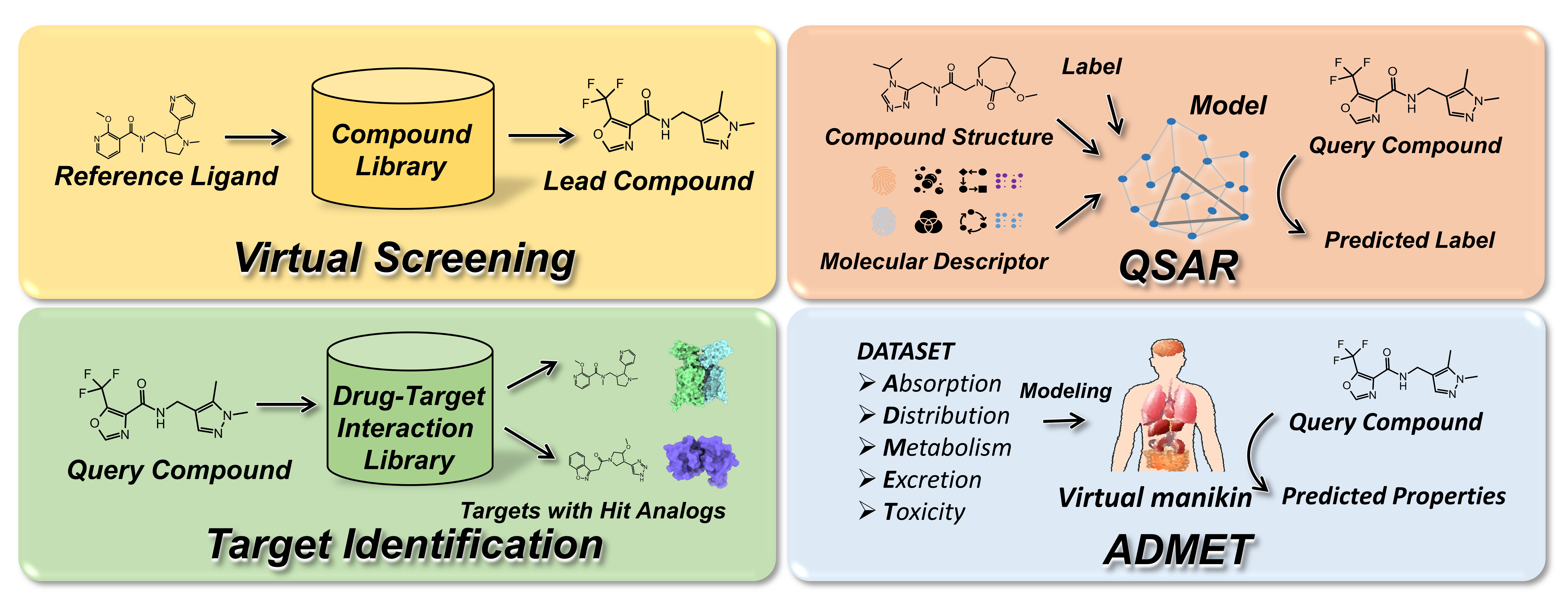
We have provided Cross-Encoder and GeminiMol models that can be used directly for inference. Here, we demonstrate the utilization of GeminiMol for virtual screening, target identification, and molecular property modeling.
Please note that while molecular fingerprints are considered simple molecular representation methods, they are an indispensable baseline (see our paper). When conducting your drug development project, we recommend exploring ECFP4, CombineFP, and GeminiMol that are provided simultaneously in our PharmProfiler.py and various molecular property modeling scripts.
In concept, molecules share similar conformational space also share similar biological activities, allowing us to predict the similarity of biological activities between molecules by comparing the similarity of GeminiMol encodings.
Here, we introduce the PharmProfiler.py, a novel approach that employs the GeminiMol encoding to establish pharmacological profiles and facilitate the search for molecules with specific properties in chemical space.
PharmProfiler.py offers the capability to conduct ligand-based virtual screening using commercially available compound libraries. Furthermore, it enables target identification through ligand similarity analysis by leveraging comprehensive drug-target relationship databases.
To support experimentation, we have included a collection of diverse commercial compound libraries and drug-target relationship databases, conveniently located in the ${geminimol_data}/compound_library/ directory.
- Prepare the pharmacological profile and compound libraries
To define a pharmacological profile, you will need to input a profile.csv file, which should have the following format:
SMILES,Label
C=CC(=O)N[C@@H]1CN(c2nc(Nc3cn(C)nc3OC)c3ncn(C)c3n2)C[C@H]1F,1.0
C=CC(=O)Nc1cccc(Nc2nc(Nc3ccc(N4CCN(C(C)=O)CC4)cc3OC)ncc2C(F)(F)F)c1,1.0
C#Cc1cccc(Nc2ncnc3cc(OCCOC)c(OCCOC)cc23)c1,1.0
COC(=O)CCC/N=C1\SCCN1Cc1ccccc1,0.4
C=C(C)[C@@H]1C[C@@H](CC2(CC=C(C)C)C(=O)C(C(CC(=O)O)c3ccccc3)=C3O[C@@H](C)[C@@H](C)C(=O)C3=C2O)C1(C)C,-0.8
C/C(=C\c1ncccc1C)[C@@H]1C[C@@H]2O[C@]2(C)CCC[C@H](C)[C@H](O)[C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1,-0.5
The "Label" column signifies the weight assigned to the reference compound. Positive values indicate that the selected compounds should bear resemblance to the reference compound, while negative values imply that the selected compounds should be dissimilar to the reference compound. Typically, positive values are assigned to active compounds, whereas negative values are assigned to inactive compounds or those causing side effects.
The compound libraries are also stored in CSV format in the ${geminimol_data}/compound_library/ directory. It is requried to maintain consistency between the SMILES column name in the profile.csv file and the compound library.
- Perform the PharmProfiler
To perform virtual screening, the following command can be used.
Here, profile_set represents the provided pharmacological profile by the user, keep_top indicates the number of compounds to be outputted in the end, and probe_cluster determines whether compounds with the same weight should be treated as a cluster. Compounds within the same cluster will be compared individually with the query mol, and the highest similarity score will be taken as the score of query mol.
We have provided a processed version of the commercial Specs and ChemDiv compound library at the ${geminimol_data}/compound_library/specs.csv and ${geminimol_data}/compound_library/ChemDiv.csv, which contained 335,212 and 1,755,930 purchasable compounds.
export job_name="Virtual_Screening"
export profile_set="profile.csv" # SMILES (same to compound library) and Label (requried)
export compound_library="${geminimol_data}/compound_library/ChemDiv.csv"
export smiles_column="SMILES" # Specify the column name in the compound_library
export weight_column="Label" # weights for profiles
export keep_top=1000
export probe_cluster="Yes"
python -u ${geminimol_app}/PharmProfiler.py "${geminimol_lib}/GeminiMol" "${job_name}" "${smiles_column}" "${compound_library}" "${profile_set}:${weight_column}" "${keep_top}" "${probe_cluster}"
To perform target identification, the compound library can be replaced with the ${geminimol_data}/compound_library/DTIDB.csv, which contains drug-target relationships. This is a processed version of the BindingDB database, which contains 2,159,221 target-ligand paris.
export job_name="Target_Identification"
export profile_set="profile.csv" # Ligand_SMILES (same to compound library), and Label (requried)
export compound_library="${geminimol_data}/compound_library/DTIDB.csv"
export smiles_column="SMILES" # Specify the column name in the compound_library
export weight_column="Label" # weights for profiles
export keep_top=2000
export probe_cluster="No"
python -u ${geminimol_app}/PharmProfiler.py "${geminimol_lib}/GeminiMol" "${job_name}" "${smiles_column}" "${compound_library}" "${profile_set}:${weight_column}" "${keep_top}" "${probe_cluster}"
After the initial run of PharmProfiler, a extracted GeminiMol feature file will be generated in the ${geminimol_data}/compound_library/. Subsequent screening tasks on the same compound library can benefit from PharmProfiler automatically reading the feature file, which helps to accelerate the running speed.
- Prepare your datasets
Before conducting molecular property modeling, it is crucial to carefully prepare your data, which includes compound structure pre-processing and dataset splitting.
Firstly, you need to clarify the chirality and protonation states of molecules in the dataset, which can be done using chemical informatics tools such as RDKit or Schrödinger software package. Typically, omitting pre-processing will not result in an error, but it may potentially impair the performance of GeminiMol.
The processed data should be saved in CSV file format, containing at least one column for SMILES and one column for Labels. Subsequently, utilize the following command for skeleton splitting. You can modify the script to change the splitting ratio, where by default, 70% of the dataset is used for training and 30% for validation and testing.
export dataset_path="data.csv"
export dataset_name="My_QSAR"
export smiles_column="SMILES" # Specify the column name in datasets
export label_column="Label" # Specify the column name in datasets
python -u ${geminimol_app}/utils/dataset_split.py "${dataset_path}" "${dataset_name}" "${smiles_column}" "${label_column}"
mkdir ${dataset_name}
mv ${dataset_name}_scaffold_*.csv ${dataset_name}/
export task=${dataset_name}
- Training the molecular property prediction model
We have presented three approaches for molecular property modeling, namely AutoQSAR (broad applicability, slow speed), PropDecoder (fast speed), and FineTuning (optimal performance, moderate speed).
Given that you have enough experience with hyperparameter tuning, the attainment of optimal performance can be accomplished through the utilization of the FineTuning script to invoke GeminiMol. Also, AutoQSAR is recommended if you lack experience with hyperparameter tuning.
2.1 Fine-Tuning on downstream task
export task="Your_Dataset" # Specify a path to your datasets (train, valid, and test)
export smiles_column="SMILES" # Specify the column name in datasets
export label_column="Label" # Specify the column name in datasets
python -u ${geminimol_app}/FineTuning.py "${task}" "${geminimol_lib}/GeminiMol" "${smiles_column}" "${label_column}" "${task}_GeminiMol"
2.2 AutoQSAR (AutoGluon)
It is recommended to try using AutoQSAR to call CombineFP or GeminiMol when you lack deep learning experience, which usually produces a model with good performance.
export encoder_method="${geminimol_lib}/GeminiMol" # only GeminiMol
In our paper, we introduced a powerful joint molecular fingerprint baseline method named CombineFP.In our experiments, the performance of CombineFP in molecular property modeling is very superior and we highly recommend trying CombineFP along with GeminiMol.
export encoder_method="ECFP4:AtomPairs:TopologicalTorsion:FCFP6" # CombineFP
Having defined the encoder, you can train the model to convert the encoding of the molecule into properties using AutoQSAR. In fact, a potential advantage of this over FineTuning is that it can decode diverse molecular properties based on the fixed encoding, which will speed up the efficiency of chemical space searching.
export task="Your_Dataset" # Specify a path to your datasets (train, valid, and test)
export smiles_column="SMILES" # Specify the column name in datasets
export label_column="Label" # Specify the column name in datasets
python -u ${geminimol_app}/AutoQSAR.py "${task}" "${encoder_method}" "${smiles_column}" "${label_column}" "" "${task}_GeminiMol"
If the integration of molecular fingerprints and a pre-trained GeminiMol model is desired for training a molecular property prediction model, either PropDecoder or AutoQSAR can be employed.
export fingerprints="ECFP4:AtomPairs:TopologicalTorsion:FCFP6:MACCS" # CombineFP+MACCS
export encoder_method="${geminimol_lib}/GeminiMol:${fingerprints}" # CombineFP+MACCS+GeminiMol
export task="Your_Dataset" # Specify a path to your datasets (train, valid, and test)
export smiles_column="SMILES" # Specify the column name in datasets
export label_column="Label" # Specify the column name in datasets
python -u ${geminimol_app}/AutoQSAR.py "${task}" "${encoder_method}" "${smiles_column}" "${label_column}" "" "${task}_GMFP"
2.3 PropDecoder
For the most tasks, performing fine-tuning or using AutoQSAR will give pretty good performance in molecular property modeling, so you don't need to try PropDecoder unless the first two give poor performance.
export task="Your_Dataset" # Specify a path to your datasets (train, valid, and test)
export smiles_column="SMILES" # Specify the column name in datasets
export label_column="Label" # Specify the column name in datasets
python -u ${geminimol_app}/PropDecoder.py "${task}" "${encoder_method}" "${smiles_column}" "${label_column}" "${task}_GeminiMol"
- Make predictions (only for AutoQSAR or fine-Tuned models)
Next, we can load the model trained based on AutoQSAR and FineTuning to predict molecular properties in a new dataset.
export model_path="QSAR_GeminiMol" # ${task}_GeminiMol when your build QSAR model
export encoder_method="${geminimol_lib}/GeminiMol" # Match to the encoders selected during QSAR model training
export extrnal_data="dataset.csv" # must contain the ${smiles_column}
export smiles_column="SMILES" # Specify the column name in datasets
export model_type="FineTuning" # FineTuning, PropDecoder, ['LightGBM', 'LightGBMLarge', 'LightGBMXT', 'NeuralNetTorch'] for AutoQSAR
python -u ${geminimol_app}/PropPredictor.py "${model_path}" "${encoder_method}" "${extrnal_data}" "${smiles_column}" "${model_type}"
If you have constructed a regression model using AutoQSAR, refer to the following command.
export model_path="QSAR_GeminiMol" # ${task}_GeminiMol when your build QSAR model
export encoder_method="${geminimol_lib}/GeminiMol" # Match to the encoders selected during QSAR model training
export extrnal_data="dataset.csv" # must contain the ${smiles_column}
export smiles_column="SMILES" # Specify the column name in datasets
export model_type="NeuralNetTorch" # ['LightGBM', 'LightGBMLarge', 'LightGBMXT', 'NeuralNetTorch'] for AutoQSAR
export task_type="regression"
python -u ${geminimol_app}/PropPredictor.py "${model_path}" "${encoder_method}" "${extrnal_data}" "${smiles_column}" "${model_type}" "${task_type}"
You can use GeminiMol to cluster molecules just like molecular fingerprints!
export encoder_method="${geminimol_lib}/GeminiMol" # Match to the encoders selected during QSAR model training
export data_table="dataset.csv" # must contain the ${smiles_column}
export smiles_column="SMILES" # Specify the column name in datasets
export output_fn="Cluster"
export cluster_num=10 # only for supervised clustering algorithm, such as K-Means
python -u ${geminimol_app}/Analyzer.py "${data_table}" "${encoder_method}" "${smiles_column}" "${output_fn}" "cluster:${cluster_num}"
You can use GeminiMol or molecular fingerprints to extract molecular features for further analysis.
export encoder_method="${geminimol_lib}/GeminiMol" # Match to the encoders selected during QSAR model training
export data_table="dataset.csv" # must contain the ${smiles_column}
export smiles_column="SMILES" # Specify the column name in datasets
export output_fn="${data_table%%.*}_Encoding"
python -u ${geminimol_app}/Analyzer.py "${data_table}" "${encoder_method}" "${smiles_column}" "${output_fn}" "encode"
Here, we present the reproducible code for training the Cross-Encoder and GeminiMol models based on the CSS descriptors of 39,290 molecules described in the paper.
Download all datasets via Zenodo for training, test and benchmark
cd ${geminimol_data}
wget https://zenodo.org/records/10450788/files/css_library.zip # only for reproducing GeminiMol training
wget https://zenodo.org/records/10450788/files/Benchmark_DUD-E.zip # only for reproducing benchmark
wget https://zenodo.org/records/10450788/files/Benchmark_LIT-PCBA.zip # only for reproducing benchmark
wget https://zenodo.org/records/10450788/files/Benchmark_QSAR.zip # only for reproducing benchmark
wget https://zenodo.org/records/10450788/files/Benchmark_TIBD.zip # only for reproducing benchmark
wget https://zenodo.org/records/10450788/files/Chem_SmELECTRA.zip # only for reproducing cross-encoder baseline
Training the Cross-Encoder
conda activate GeminiMol
export model_name="CrossEncoder"
export batch_size_per_gpu=200 # batch size = 200 (batch_size_per_gpu) * 4 (gpu number)
export epoch=20 # max epochs
export lr="1.0e-3" # learning rate
export label_list="MCMM1AM_MAX:LCMS2A1Q_MAX:MCMM1AM_MIN:LCMS2A1Q_MIN" # ShapeScore:ShapeAggregation:ShapeOverlap:CrossSim:CrossAggregation:CrossOverlap
CUDA_VISIBLE_DEVICES=0,1,2,3 python ${geminimol_app}/CrossEncoder_Training.py "${geminimol_data}/css_library/" "${geminimol_data}/Chem_SmELECTRA" "${epoch}" "${lr}" "${batch_size_per_gpu}" "${model_name}" "${geminimol_data}/benchmark.json" "${label_list}"
Training the GeminiMol Encoder and Decoder of CSS descriptors
conda activate GeminiMol
export model_name="GeminiMol"
export batch_size=512
export epoch=20 # max epochs
export patience=50 # for early stoping
export GNN='WLN' # Weisfeiler-Lehman Network (WLN)
export network="MeanMLP:2048:4:2048:None:0:5:0"
export label_dict="ShapeScore:0.2,ShapeAggregation:0.2,ShapeOverlap:0.05,ShapeDistance:0.05,CrossSim:0.15,CrossAggregation:0.15,CrossDist:0.05,CrossOverlap:0.05,MCS:0.1"
CUDA_VISIBLE_DEVICES=0 python -u ${geminimol_app}/GeminiMol_Training.py "${geminimol_data}/css_library/" "${epoch}" "${batch_size}" "${GNN}" "${network}" "${label_dict}" "${model_name}" "${patience}" "${geminimol_data}/benchmark.json"
Additionally, benchmark test scripts were provided. With this code, the community can reproduce the results reported in the paper, explore different model architectures, even incorporate additional molecular similarity data to further enhance the performance of the models.
Benchmarking molecular fingerprints and GeminiMol on virutual screening and target identification
For each molecular fingerprint, we used all supported similarity metrics, including Tanimoto, Cosine, and Tversky. For the GeminiMol model, in addition to the projected heads used in pre-training, we introduced similarities between molecular representation vectors, including Cosine and Pearson. It is worth noting that in practice we cannot be sure which combination of molecular fingerprints and similarity metrics is optimal, and therefore each combination is considered an independent method in benchmarking.
conda activate GeminiMol
# benchmarking Fixed GeminiMol models and Fingerprints
for task in "DUDE" "LIT-PCBA" "TIBD" # zero-shot tasks
do
for model_name in "FCFP6" "MACCS" "RDK" "ECFP6" "FCFP4" \
"TopologicalTorsion" "AtomPairs" "ECFP4" \
"${geminimol_lib}/GeminiMol"
do
mkdir -p ${model_name}
CUDA_VISIBLE_DEVICES=0 python -u ${geminimol_app}/benchmark.py "${model_name}" "${geminimol_data}/benchmark.json" "${task}"
done
done
Benchmarking molecular fingerprints and GeminiMol on molecular property modeling
It is worth noting that different decoders exhibit varying performance on different tasks and encodings. Therefore, it is essential to select the appropriate decoder for each specific molecular encoder and task. In practice, we can determine when the model should stop-training and choose the optimal decoder architecture by dividing the training, validation and test sets. Consequently, all results should be merged using a data pivot table to analyze the optimal decoder for each encoder-task combination. In our work, the hyperparameters of the PropDecoder were chosen based on empirical experience and were not subjected to any hyperparameter tuning. Performing further hyperparameter tuning for each task may potentially yield improved performance.
for task in "ADMET-C" "ADMET-R" \
"LIT-QSAR" "CELLS-QSAR" "ST-QSAR" "PW-QSAR" \
"PropDecoder-ADMET" "PropDecoder-QSAR" # fixed the molecular encoder
do
for model_name in "CombineFP" \
"FCFP6" "MACCS" "RDK" "ECFP6" "FCFP4" "TopologicalTorsion" "AtomPairs" "ECFP4" \
"${geminimol_lib}/GeminiMol"
do
mkdir -p ${model_name}
CUDA_VISIBLE_DEVICES=0 python -u ${geminimol_app}/benchmark.py "${model_name}" "${geminimol_data}/benchmark.json" "${task}"
done
done
for task in "FineTuning-ADMET" "FineTuning-QSAR"; do # benchmarking with FineTuning GeminiMol models
for model_name in "${geminimol_lib}/GeminiMol"; do
CUDA_VISIBLE_DEVICES=0 python -u ${geminimol_app}/benchmark.py "${model_name}" "${geminimol_data}/benchmark.json" "${task}"
done
done
Conformational Space Profiling Enhances Generic Molecular Representation for AI-Powered Ligand-Based Drug Discovery
Lin Wang, Shihang Wang, Hao Yang, Shiwei Li, Xinyu Wang, Yongqi Zhou, Siyuan Tian, Lu Liu, Fang Bai
Advanced Science, 2024; doi: 10.1002/advs.202403998
GeminiMol is released under the Academic Free Licence, which permits academic use, modification and distribution free of charge. GeminiMol can be utilized in academic publications, open-source software projects, and open-source competitions (e.g. Kaggle competitions under the MIT Open Source license).
GeminiMol prohibits unauthorised commercial use, including commercial training and as part of a paid computational platform, which intended to prevent speculators from exploiting informational asymmetry for profit. Communication and authorization with our supervisor is permitted for its application in pipeline development and research activities within pharmaceutical R&D.
We welcome community contributions of extension tools based on the GeminiMol model, etc. If you have any questions not covered in this overview, please contact the GeminiMol Developer Team. We would like to hear your feedback and understand how GeminiMol has been useful in your research. Share your stories with us.
We appreciate the technical support provided by the engineers of the high-performance computing cluster of ShanghaiTech University. Lin Wang also thanks Jianxin Duan, Gaokeng Xiao, Quanwei Yu, Zheyuan Shen, Shenghao Dong, Huiqiong Li, Zongquan Li, and Fenglei Li for providing technical support, inspiration and help for this work. We express our gratitude to Dr. Zhongji Pu, Dr. Quanwei Yu for their invaluable assistance in third-party testing for model installation, reproducibility and application.
We also thank the developers and maintainers of MarcoModel and PhaseShape modules in the Schrödinger package. Besides, GeminiMol communicates with and/or references the following separate libraries and packages, we thank all their contributors and maintainers!